Molecular docking
This is the main protocol of Jupyter Dock.
Content of this notebook
Feching system and cleanup
Protein and ligand sanitization
System Visualization
Docking with AutoDock Vina
Receptor preparation
Ligand preparation
Docking box definition
Docking
PDBQT results file conversion to SDF
3D visualization of docking results
2D interaction table and map
Docking with Smina
Receptor preparation
Ligand preparation
Docking box definition
Docking
3D visualization of docking results
2D interaction table and map
Docking with LeDock
Receptor preparation
Ligand preparation
Docking box definition
Docking
DOK results file conversion to SDF
3D visualization of docking results
2D interaction table and map
[1]:
from pymol import cmd
import py3Dmol
from vina import Vina
from openbabel import pybel
from rdkit import Chem
from rdkit.Chem import AllChem, Draw
from meeko import MoleculePreparation
from meeko import obutils
import MDAnalysis as mda
from MDAnalysis.coordinates import PDB
import prolif as plf
from prolif.plotting.network import LigNetwork
import sys, os
sys.path.insert(1, 'utilities/')
from utils import fix_protein, getbox, generate_ledock_file, pdbqt_to_sdf, dok_to_sdf
import warnings
warnings.filterwarnings("ignore")
%config Completer.use_jedi = False
It’s a good idea to run the test protocols before attempting custom projects. Later, the user can specify the location of his/her project and save all of the files separately.
[2]:
os.chdir('test/Molecular_Docking/')
1. Feching system and cleanup
Implementing Pymol is a simple way to download PDB structures. The user can launch this or any other Jupyter Dock’s protocol by providing his or her own files.
[3]:
cmd.fetch(code='1AZ8',type='pdb1')
cmd.select(name='Prot',selection='polymer.protein')
cmd.select(name='Lig',selection='organic')
cmd.save(filename='1AZ8_clean.pdb',format='pdb',selection='Prot')
cmd.save(filename='1AZ8_lig.mol2',format='mol2',selection='Lig')
cmd.delete('all')
2. Protein and ligand sanitization
2.1. Protein Sanitization
Method 1: LePro
The Lephar molecular docking suite includes a very powerful tool for automatically preparing proteins for molecular docking. As a result, for these protocols, it will be the preferred tool for protein preparation. The protein prepared by LePro can be used in AutoDock VIna and LeDock.
[4]:
!../../bin/lepro_linux_x86 # Launch this cell to see parameters
************************************************************
* LePro *
* Add hydrogen atoms to a protein & *
* write the input file for LeDock *
* Copyright (C) 2013-14 Hongtao Zhao, PhD *
* Email: htzhaovv@gmail.com *
************************************************************
----------Usage:
lepro [PDB file] [-rot || -metal || -p]
-rot [[chain] resid] align principal axes of the binding site with Cartesian
-metal keep ZN/MN/CA/MG
-metal -p redistribute metal charge to protein
[5]:
!../../bin/lepro_linux_x86 {'1AZ8_clean.pdb'}
os.rename('pro.pdb','1AZ8_clean_H.pdb') # Output from lepro is pro.pdb, this line will change the name to '1AZ8_clean_H.pdb'
Method 2: fix_protein (PDBFixer)
For proteins with missing amino acids or residues, or to ensure a more thorough sanitization of protein Jupyter Dock includes the fix_protein() function, which employs PDBFixer to correct a wide range of common errors in protein pdb files. Furthermore, PDBFixer enables the assignment of pH-dependent protonation states to proteins.
Warning 1: It is possible to encounter problems with protein fixing when using fix_protein() and AutoDock Tools’ prepare_receptor or when running LeDock. To resolve the issue, it is best to set the parameter -A hydrogens in prepare receptor.
Hint: PDBFixer is a great solution for many systems because it can solve serious problems in PDB files. As a result, PDBFixer renumbers the residues beginning with 1 regardless of the numbering on the original PDB file. To address this issue, the fix_protein() function includes a protocol for atomically renumbering the residues in accordance with the original PDB file.
fix_protein ( params )
Params:
filename: str or path-like ; input file containing protein struture to be modified, file extrension must be pdb
addHs_pH: float ; Add hydrogens at user defined pH
try_renumberResidues: bool ; By default PDBFixer renumarets residues starting in 1, this option tries to recover originar residues numbering
output: str or path-like ; output filename, extension must be pdb
fix_protein(filename='1AZ8_clean.pdb',addHs_pH=7.4,try_renumberResidues=True,output='1AZ8_clean_H.pdb')
2.2. Ligand sanitization
Due to the variability of ligands and formats, ligand sanitization and preparation can be one of the most difficult tasks to complete. Setting protonation states for a ligand, for example, can be difficult. It is highly recommended that the user knows and understands the proper states for his/her ligand(s) when using Jupyter Dock or any other molecular docking approach.
In this example, after splitting the ligand and protein after fetching with pymol, the ligand has sanitization problems (sanitize=False in Chem.MolFromMol2File)
[6]:
m=Chem.MolFromMol2File('1AZ8_lig.mol2',sanitize=False)
Draw.MolToImage(m)
RDKit WARNING: [16:39:50] 1AZ8: Warning - no explicit hydrogens in mol2 file but needed for formal charge estimation.
[6]:
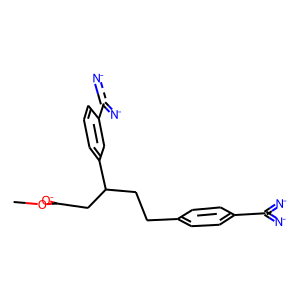
Hint: One solution for simple sanitization problems is to use OpenBAbel to convert the molecule and add the necessary hydrogens for molecular docking (Pybel). The definitions for carrying out molecules in OpenBabel differ from those in RDKIt. As a result, OpenBabel is capable of handling the conversion.
[7]:
mol= [m for m in pybel.readfile(filename='1AZ8_lig.mol2',format='mol2')][0]
mol.addh()
out=pybel.Outputfile(filename='1AZ8_lig_H.mol2',format='mol2',overwrite=True)
out.write(mol)
out.close()
The end result of ligand sanitization is a new molecule that RDKit can display without having to use the sanitization parameter. Furthermore, the output structure for this example corresponds exactly to the one reported in the PDB database (PDB 1AZ8)
[8]:
m=Chem.MolFromMol2File('1AZ8_lig_H.mol2')
m
[8]:
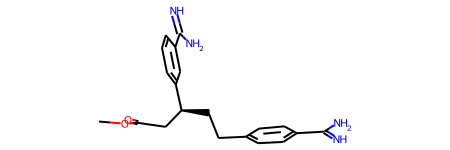
3. System Visualization
A cool feature of Jupyter Dock is the posibility of visualize ligand-protein complexes and docking results into the notebbok. All this thanks to the powerful py3Dmol.
Now the protein and ligand have been sanitized it would be recomended to vissualize the ligand-protein reference system.
[9]:
view = py3Dmol.view()
view.removeAllModels()
view.setViewStyle({'style':'outline','color':'black','width':0.1})
view.addModel(open('1AZ8_clean_H.pdb','r').read(),format='pdb')
Prot=view.getModel()
Prot.setStyle({'cartoon':{'arrows':True, 'tubes':True, 'style':'oval', 'color':'white'}})
view.addSurface(py3Dmol.VDW,{'opacity':0.6,'color':'white'})
view.addModel(open('1AZ8_lig_H.mol2','r').read(),format='mol2')
ref_m = view.getModel()
ref_m.setStyle({},{'stick':{'colorscheme':'greenCarbon','radius':0.2}})
view.zoomTo()
view.show()
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension:
jupyter labextension install jupyterlab_3dmol
4. Docking with AutoDock Vina
AutoDock Vina (Vina) is one of the docking engines in the AutoDock Suite, together with AutoDock4 (AD4), AutoDockGPU, AutoDockFR, and AutoDock-CrankPep, and arguably among the most widely used and successful docking engines. The reasons for this success are mostly due to its ease of use and its speed (up to 100x faster than AD4), when compared to the other docking engines in the suite and elsewhere, as well as being open source.
4.1. Protein preparation
After sanitization, the protein docking preparation includes converting it to the PDBQT file format, which stores the atomic coordinates, partial charges, and AutoDock atom types for both the receptor and the ligand.
Hint: Despite the fact that the PDBQT format includes changes (Q) and atom types (T) for molecular docking. The charges are not required for Autodock Vina, which computes electrostatic interactions using its own force field. Although, when using AutoDock instead of AutoDock Vina, the Q term is required. More information can be found here:https://autodock-vina.readthedocs.io/en/latest/introduction.html
Method 1: AutoDock Tools prepare_receptor
The AutoDock Tools are the best way to prepare the receptor for AutoDock Vina. Nonetheless, AutoDock Tools is a comprehensive suite of programs and scripts that are difficult to manage. Jupyter Dock execute the prepare receptor and prepare_ligand functions on their own. As a result, obtaining proper PDBQT files for AutoDock Vina has never been easier.
Warning: There is currently only one method for preparing a PDBQT file for a receptor. It is expected that new methods apart from AutoDock Tools executables will be included in the near future into Jupyter Dock.
[10]:
!../../bin/prepare_receptor #Lauch this cell to see parameters
prepare_receptor4: receptor filename must be specified.
Usage: prepare_receptor4.py -r filename
Description of command...
-r receptor_filename
supported file types include pdb,mol2,pdbq,pdbqs,pdbqt, possibly pqr,cif
Optional parameters:
[-v] verbose output (default is minimal output)
[-o pdbqt_filename] (default is 'molecule_name.pdbqt')
[-A] type(s) of repairs to make:
'bonds_hydrogens': build bonds and add hydrogens
'bonds': build a single bond from each atom with no bonds to its closest neighbor
'hydrogens': add hydrogens
'checkhydrogens': add hydrogens only if there are none already
'None': do not make any repairs
(default is 'None')
[-C] preserve all input charges ie do not add new charges
(default is addition of gasteiger charges)
[-p] preserve input charges on specific atom types, eg -p Zn -p Fe
[-U] cleanup type:
'nphs': merge charges and remove non-polar hydrogens
'lps': merge charges and remove lone pairs
'waters': remove water residues
'nonstdres': remove chains composed entirely of residues of
types other than the standard 20 amino acids
'deleteAltB': remove XX@B atoms and rename XX@A atoms->XX
(default is 'nphs_lps_waters_nonstdres')
[-e] delete every nonstd residue from any chain
'True': any residue whose name is not in this list:
['CYS','ILE','SER','VAL','GLN','LYS','ASN',
'PRO','THR','PHE','ALA','HIS','GLY','ASP',
'LEU', 'ARG', 'TRP', 'GLU', 'TYR','MET',
'HID', 'HSP', 'HIE', 'HIP', 'CYX', 'CSS']
will be deleted from any chain.
NB: there are no nucleic acid residue names at all
in the list and no metals.
(default is False which means not to do this)
[-M] interactive
(default is 'automatic': outputfile is written with no further user input)
[-d dictionary_filename] file to contain receptor summary information
[-w] assign each receptor atom a unique name: newname is original name plus its index(1-based)
[11]:
!../../bin/prepare_receptor -v -r {'1AZ8_clean_H.pdb'} -o {'1AZ8_clean_H.pdbqt'}
set verbose to True
set receptor_filename to 1AZ8_clean_H.pdb
set outputfilename to 1AZ8_clean_H.pdbqt
read 1AZ8_clean_H.pdb
setting up RPO with mode= automatic and outputfilename= 1AZ8_clean_H.pdbqt
charges_to_add= gasteiger
delete_single_nonstd_residues= None
adding gasteiger charges to peptide
4.2. Ligand preparation
As previously discussed, one of the major challenges for proper docking experimentation is ligand preparation.
Hint: The hydroges in this example were set during the ligand sanitization step (section 2.2). As a result, the ligand will be prepared as is. Furthermore, before running the docking, it is highly recommended to inspect the ligand(s) after preparation to ensure proper structure and molecule features.
Method 1: AutoDock Tools prepare_ligand
[12]:
!../../bin/prepare_ligand #Launch this cell to see parameters
prepare_ligand4: ligand filename must be specified.
Usage: prepare_ligand4.py -l filename
Description of command...
-l ligand_filename (.pdb or .mol2 or .pdbq format)
Optional parameters:
[-v] verbose output
[-o pdbqt_filename] (default output filename is ligand_filename_stem + .pdbqt)
[-d] dictionary to write types list and number of active torsions
[-A] type(s) of repairs to make:
bonds_hydrogens, bonds, hydrogens (default is to do no repairs)
[-C] do not add charges (default is to add gasteiger charges)
[-p] preserve input charges on an atom type, eg -p Zn
(default is not to preserve charges on any specific atom type)
[-U] cleanup type:
nphs_lps, nphs, lps, '' (default is 'nphs_lps')
[-B] type(s) of bonds to allow to rotate
(default sets 'backbone' rotatable and 'amide' + 'guanidinium' non-rotatable)
[-R] index for root
[-F] check for and use largest non-bonded fragment (default is not to do this)
[-M] interactive (default is automatic output)
[-I] string of bonds to inactivate composed of
of zero-based atom indices eg 5_13_2_10
will inactivate atoms[5]-atoms[13] bond
and atoms[2]-atoms[10] bond
(default is not to inactivate any specific bonds)
[-Z] inactivate all active torsions
(default is leave all rotatable active except amide and guanidinium)
[-g] attach all nonbonded fragments
[-s] attach all nonbonded singletons:
NB: sets attach all nonbonded fragments too
(default is not to do this)
[-w] assign each ligand atom a unique name: newname is original name plus its index(1-based)
[13]:
!../../bin/prepare_ligand -v -l {'1AZ8_lig_H.mol2'} -o {'1AZ8_lig_H.pdbqt'}
set verbose to True
set ligand_filename to 1AZ8_lig_H.mol2
set outputfilename to 1AZ8_lig_H.pdbqt
read 1AZ8_lig_H.mol2
setting up LPO with mode= automatic and outputfilename= 1AZ8_lig_H.pdbqt
and check_for_fragments= False
and bonds_to_inactivate=
returning 0
No change in atomic coordinates
Method 2: Meeko
This is the preferred method out of AutoDock Tools. Apart from ligand preparation, Meeko provides tools for other docking aspects that the AutoDock Tools software suite does not cover. Hydrated docking and macrocycles are two examples. More about Meeko https://pypi.org/project/meeko/
Info To run Meeko change the next cell type from “Markdown” to “Code”.
mol = obutils.load_molecule_from_file('1AZ8_lig_H.mol2')
preparator = MoleculePreparation(merge_hydrogens=True,hydrate=False)
preparator.prepare(mol)
preparator.write_pdbqt_file('1AZ8_lig_H.pdbqt')
Method 3: Pybel
OpenBabel (pybel) can handle a variety of file formats and conversions between them. As a result, any chemical format file can be converted directly to PDBQT. Nonetheless, pybel may encounter issues if the source file format contains errors.
Info To run Pybel change the next cell type from “Markdown” to “Code”.
ligand = [m for m in pybel.readfile(filename='1AZ8_lig_H.mol2',format='mol2')][0]
out=pybel.Outputfile(filename='1AZ8_lig_H.pdbqt',format='pdbqt',overwrite=True)
out.write(ligand)
out.close()
4.3. Box definition
This is possibly the most important feature of Jupyter Dock. Making a docking box without the use of a visualizer or any other additional tools. As a result, AutoDock Vina (and LeDock) can now be run entirely in a Jupyter notebook.
Mengwu Xiao and his clever Pymol plug-in “GetBox” inspired the box definition in Jupyter Dock.
Jupyter Dock makes use of Mengwu Xiao Pymol implementation, but this time through the Pymol API.
Warning: The function get_box() is built into the Pymol API. As a result, in order to compute the docking box, the docking system files must be initialized using Pymol.
Hint: The integration of get_box() as Pymol integration allows the definition of amazing boxes based on Pymol’s powerful selection tools. A ligand, a residue, a set of residues, atom(s), pseudoatom(s), and any custom selection valid within the Pymol selection algebra can be used to set the box.
get_box( params )
params:
selection: str , pymol_selection; The selection for docking box, can be atom, resn, resid, or any other pymol selection
extending: float; value to extend the boundaries of the selection
software: str , ‘vina’,’ledock’, ‘both’ ; Depending on selected software the funtion will provide the box coordinates in vina, ledock, or both formats
[14]:
cmd.load(filename='1AZ8_clean_H.pdb',format='pdb',object='prot')
cmd.load(filename='1AZ8_lig_H.mol2',format='mol2',object='lig')
center, size= getbox(selection='lig',extending=5.0,software='vina')
cmd.delete('all')
print(center,'\n',size)
{'center_x': 31.859049797058105, 'center_y': 13.347449779510498, 'center_z': 17.06589984893799}
{'size_x': 24.56949806213379, 'size_y': 18.123299598693848, 'size_z': 17.374399185180664}
4.4. Docking
AutoDock Vina 1.2.0, which was recently released, now allows AutoDock Vina to be executed using Python Bindings. Jupyter Dock takes advantage of this feature to make the docking protocol run entirely within a Jupyter notebook.
[15]:
v = Vina(sf_name='vina')
v.set_receptor('1AZ8_clean_H.pdbqt')
v.set_ligand_from_file('1AZ8_lig_H.pdbqt')
v.compute_vina_maps(center=[center['center_x'], center['center_y'], center['center_z']],
box_size=[size['size_x'], size['size_y'], size['size_z']])
'''
# Score the current pose
energy = v.score()
print('Score before minimization: %.3f (kcal/mol)' % energy[0])
# Minimized locally the current pose
energy_minimized = v.optimize()
print('Score after minimization : %.3f (kcal/mol)' % energy_minimized[0])
v.write_pose('1iep_ligand_minimized.pdbqt', overwrite=True)
'''
# Dock the ligand
v.dock(exhaustiveness=10, n_poses=10)
v.write_poses('1AZ8_lig_vina_out.pdbqt', n_poses=10, overwrite=True)
4.5. PDBQT results file conversion to SDF
Because of the unique characteristics of the PDBQT format, it may be difficult to visualize the results or perform other analyses after docking. As a result, Jupyter Dock automatically converts the PDFBQT format to the more common SDF format. The file conversion preserves the chemical properties of the compounds after they have been sanitized and prepared. Furthermore, the “Pose” and “Score” information is saved as a molecule attribute. Such data can be accessed directly in the file or via Pybel or RDKit.
pdbqt_to_sdf ( params )
params:
pdbqt_file: str or path-like string ; pdbqt file to be converted, extension must be pdbqt
output: str or path-like string ; output sdf file, extension must be sdf
[16]:
pdbqt_to_sdf(pdbqt_file='1AZ8_lig_vina_out.pdbqt',output='1AZ8_lig_vina_out.sdf')
4.6. 3D visualization of docking results (AutoDock Vina)
As with the system visualization (section 3), the docking results can be inspected and compared to the reference structure (if exist). The ligand’s “Pose” and “Score” information will also be displayed to show how acces to this molecule’s attributes.
[17]:
view = py3Dmol.view()
view.removeAllModels()
view.setViewStyle({'style':'outline','color':'black','width':0.1})
view.addModel(open('1AZ8_clean_H.pdb','r').read(),format='pdb')
Prot=view.getModel()
Prot.setStyle({'cartoon':{'arrows':True, 'tubes':True, 'style':'oval', 'color':'white'}})
view.addSurface(py3Dmol.VDW,{'opacity':0.6,'color':'white'})
view.addModel(open('1AZ8_lig_H.mol2','r').read(),format='mol2')
ref_m = view.getModel()
ref_m.setStyle({},{'stick':{'colorscheme':'magentaCarbon','radius':0.2}})
results=Chem.SDMolSupplier('1AZ8_lig_vina_out.sdf')
p=Chem.MolToMolBlock(results[0],False)
print('Reference: Magenta | Vina Pose: Cyan')
print ('Pose: {} | Score: {}'.format(results[0].GetProp('Pose'),results[0].GetProp('Score')))
view.addModel(p,'mol')
x = view.getModel()
x.setStyle({},{'stick':{'colorscheme':'cyanCarbon','radius':0.2}})
view.zoomTo()
view.show()
Reference: Magenta | Vina Pose: Cyan
Pose: 1 | Score: -7.746
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension:
jupyter labextension install jupyterlab_3dmol
4.7. 2D interaction table and map
Inspecting the molecular interactions is one of the most common analyses performed by a computational scientist following a docking experiment. As a result, Jupyter Dock uses ProLif to create a table of ligand-protein molecular interactions as well as the corresponding 2D map.
Info: ProLif uses RDKit and MDAnalysis to map the molecular interaction between a ligand and a protein. As a result, some protein preparations, including the use of LePro, can result in errors that impede analysis. Jupyter Dock’s _**fix protein()_** function can be used to avoid such errors and provide a suitable protein structure for the analysis.
[18]:
fix_protein(filename='1AZ8_clean.pdb',addHs_pH=7.4,try_renumberResidues=True,output='1AZ8_clean_H_fix.pdb')
Warning: importing 'simtk.openmm' is deprecated. Import 'openmm' instead.
[19]:
# load protein
prot = mda.Universe("1AZ8_clean_H_fix.pdb")
prot = plf.Molecule.from_mda(prot)
prot.n_residues
# load ligands
lig_suppl = list(plf.sdf_supplier('1AZ8_lig_vina_out.sdf'))
# generate fingerprint
fp = plf.Fingerprint()
fp.run_from_iterable(lig_suppl, prot)
results_df = fp.to_dataframe(return_atoms=True)
results_df
[19]:
| ligand | UNL1 | |||
|---|---|---|---|---|
| protein | ASP189.A | CYS191.A | CYS42.A | GLY219.A |
| interaction | HBDonor | Hydrophobic | Hydrophobic | HBDonor |
| Frame | ||||
| 0 | (None, None) | (None, None) | (None, None) | (9, 0) |
| 1 | (11, 0) | (15, 0) | (None, None) | (12, 0) |
| 2 | (None, None) | (None, None) | (None, None) | (None, None) |
| 3 | (None, None) | (None, None) | (None, None) | (9, 0) |
| 4 | (None, None) | (None, None) | (None, None) | (None, None) |
| 5 | (None, None) | (None, None) | (None, None) | (9, 0) |
| 6 | (None, None) | (None, None) | (None, None) | (23, 0) |
| 7 | (None, None) | (None, None) | (7, 0) | (None, None) |
| 8 | (25, 0) | (None, None) | (None, None) | (26, 0) |
| 9 | (None, None) | (None, None) | (None, None) | (23, 0) |
[20]:
net = LigNetwork.from_ifp(results_df,lig_suppl[0],kind="frame", frame=0,rotation=270)
net.display()
[20]:
5. Docking with Smina
Despite the presence of Python bindings in AutoDock Vina 1.2.0, other tools that incorporate AutoDock Vina allow for cool features such as custom score functions (smina), fast execution (qvina), and the use of wider boxes (qvina-w). Jupyter Dock can run such binaries in a notebook, giving users more options.
Smina is a fork of AutoDock Vina that is customized to better support scoring function development and high-performance energy minimization. Smina is maintained by David Koes at the University of Pittsburgh and is not directly affiliated with the AutoDock project.
Info: The following cell contains an example of using Smina to run the current docking example. However, the executable files for qvina and qvina-w are available in the Jupyter Dock repo’s bin directory. As a result, the user can use such a tool by adding the necessary cells or replacing the current docking engine.
5.1. Receptor preparation
Despite the fact that Smina is a modified version of AutoDock Vina, the input file for a receptor in Smina can be either a PDBQT file or a PDB file with explicit hydrogens in all residues. At this point, we can make use of the file sanitization steps as well as a protein structure from Jupyter Dock’s fix_protein() function.
5.2. Ligand preparation
In Smina, we can use any OpenBabel format for ligand input and docking results, just like we can for receptor preparation. As a result, after sanitization, we can use the ligand MOL2 file here.
5.3. Docking box definition
This step can be completed in the same manner as the AutoDock Vina box definition.
5.4. Docking
Jupyter Dock comes with the smina executable for Linux and Mac OS. By running the binary file, the parameters can be accessed.
[21]:
!../../bin/smina #Lauch this cell to see parameters
Missing receptor.
Correct usage:
Input:
-r [ --receptor ] arg rigid part of the receptor (PDBQT)
--flex arg flexible side chains, if any (PDBQT)
-l [ --ligand ] arg ligand(s)
--flexres arg flexible side chains specified by comma
separated list of chain:resid or
chain:resid:icode
--flexdist_ligand arg Ligand to use for flexdist
--flexdist arg set all side chains within specified distance
to flexdist_ligand to flexible
Search space (required):
--center_x arg X coordinate of the center
--center_y arg Y coordinate of the center
--center_z arg Z coordinate of the center
--size_x arg size in the X dimension (Angstroms)
--size_y arg size in the Y dimension (Angstroms)
--size_z arg size in the Z dimension (Angstroms)
--autobox_ligand arg Ligand to use for autobox
--autobox_add arg Amount of buffer space to add to auto-generated
box (default +4 on all six sides)
--no_lig no ligand; for sampling/minimizing flexible
residues
Scoring and minimization options:
--scoring arg specify alternative builtin scoring function
--custom_scoring arg custom scoring function file
--custom_atoms arg custom atom type parameters file
--score_only score provided ligand pose
--local_only local search only using autobox (you probably
want to use --minimize)
--minimize energy minimization
--randomize_only generate random poses, attempting to avoid
clashes
--minimize_iters arg (=0) number iterations of steepest descent; default
scales with rotors and usually isn't sufficient
for convergence
--accurate_line use accurate line search
--minimize_early_term Stop minimization before convergence conditions
are fully met.
--approximation arg approximation (linear, spline, or exact) to use
--factor arg approximation factor: higher results in a
finer-grained approximation
--force_cap arg max allowed force; lower values more gently
minimize clashing structures
--user_grid arg Autodock map file for user grid data based
calculations
--user_grid_lambda arg (=-1) Scales user_grid and functional scoring
--print_terms Print all available terms with default
parameterizations
--print_atom_types Print all available atom types
Output (optional):
-o [ --out ] arg output file name, format taken from file
extension
--out_flex arg output file for flexible receptor residues
--log arg optionally, write log file
--atom_terms arg optionally write per-atom interaction term
values
--atom_term_data embedded per-atom interaction terms in output
sd data
Misc (optional):
--cpu arg the number of CPUs to use (the default is to
try to detect the number of CPUs or, failing
that, use 1)
--seed arg explicit random seed
--exhaustiveness arg (=8) exhaustiveness of the global search (roughly
proportional to time)
--num_modes arg (=9) maximum number of binding modes to generate
--energy_range arg (=3) maximum energy difference between the best
binding mode and the worst one displayed
(kcal/mol)
--min_rmsd_filter arg (=1) rmsd value used to filter final poses to remove
redundancy
-q [ --quiet ] Suppress output messages
--addH arg automatically add hydrogens in ligands (on by
default)
Configuration file (optional):
--config arg the above options can be put here
Information (optional):
--help display usage summary
--help_hidden display usage summary with hidden options
--version display program version
[22]:
!../../bin/smina -r {'1AZ8_clean_H.pdb'} -l {'1AZ8_lig_H.mol2'} -o {'1AZ8_lig_smina_out.sdf'} --center_x 31.859 --center_y 13.34 --center_z 17.065 --size_x 24.569 --size_y 18.12 --size_z 17.37 --exhaustiveness 8 --num_modes 10
_______ _______ _________ _ _______
( ____ \( )\__ __/( ( /|( ___ )
| ( \/| () () | ) ( | \ ( || ( ) |
| (_____ | || || | | | | \ | || (___) |
(_____ )| |(_)| | | | | (\ \) || ___ |
) || | | | | | | | \ || ( ) |
/\____) || ) ( |___) (___| ) \ || ) ( |
\_______)|/ \|\_______/|/ )_)|/ \|
smina is based off AutoDock Vina. Please cite appropriately.
Weights Terms
-0.035579 gauss(o=0,_w=0.5,_c=8)
-0.005156 gauss(o=3,_w=2,_c=8)
0.840245 repulsion(o=0,_c=8)
-0.035069 hydrophobic(g=0.5,_b=1.5,_c=8)
-0.587439 non_dir_h_bond(g=-0.7,_b=0,_c=8)
1.923 num_tors_div
Using random seed: 1899689032
0% 10 20 30 40 50 60 70 80 90 100%
|----|----|----|----|----|----|----|----|----|----|
***************************************************
mode | affinity | dist from best mode
| (kcal/mol) | rmsd l.b.| rmsd u.b.
-----+------------+----------+----------
1 -8.1 0.000 0.000
2 -7.7 4.282 7.834
3 -7.6 1.560 2.181
4 -7.4 3.914 7.467
5 -7.3 4.727 7.570
6 -7.3 3.979 7.582
7 -7.1 4.102 6.950
8 -7.1 3.272 4.506
9 -7.0 7.062 10.688
10 -7.0 5.740 9.337
Refine time 29.933
Loop time 30.808
5.6. 3D visualization of docking results
As with the system visualization (section 3), the docking results can be inspected and compared to the reference structure (if one exists). Smina saves the “minimizedAffinity” information corresponding to the docking score as the molecule’s attribute.
[23]:
view = py3Dmol.view()
view.removeAllModels()
view.setViewStyle({'style':'outline','color':'black','width':0.1})
view.addModel(open('1AZ8_clean_H.pdb','r').read(),format='pdb')
Prot=view.getModel()
Prot.setStyle({'cartoon':{'arrows':True, 'tubes':True, 'style':'oval', 'color':'white'}})
view.addSurface(py3Dmol.VDW,{'opacity':0.6,'color':'white'})
view.addModel(open('1AZ8_lig_H.mol2','r').read(),format='mol2')
ref_m = view.getModel()
ref_m.setStyle({},{'stick':{'colorscheme':'magentaCarbon','radius':0.2}})
results=Chem.SDMolSupplier('1AZ8_lig_smina_out.sdf')
p=Chem.MolToMolBlock(results[0],False)
print('Reference: Magenta | Smina Pose: Cyan')
print ('Score: {}'.format(results[0].GetProp('minimizedAffinity')))
view.addModel(p,'mol')
x = view.getModel()
x.setStyle({},{'stick':{'colorscheme':'cyanCarbon','radius':0.2}})
view.zoomTo()
view.show()
Reference: Magenta | Smina Pose: Cyan
Score: -8.10622
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension:
jupyter labextension install jupyterlab_3dmol
5.7. 2D interaction table and map
Inspecting the molecular interactions is one of the most common analyses performed by a computational scientist following a docking experiment. As a result, Jupyter Dock uses ProLif to create a table of ligand-protein molecular interactions as well as the corresponding 2D map.
Info: ProLif uses RDKit and MDAnalysis to map the molecular interaction between a ligand and a protein. As a result, some protein preparations, including the use of LePro, can result in errors that impede analysis. Jupyter Dock’s _**fix protein()_** function can be used to avoid such errors and provide a suitable protein structure for the analysis.
[24]:
# load protein
prot = mda.Universe("1AZ8_clean_H_fix.pdb")
prot = plf.Molecule.from_mda(prot)
prot.n_residues
# load ligands
lig_suppl = list(plf.sdf_supplier('1AZ8_lig_smina_out.sdf'))
# generate fingerprint
fp = plf.Fingerprint()
fp.run_from_iterable(lig_suppl, prot)
results_df = fp.to_dataframe(return_atoms=True)
results_df
[24]:
| ligand | UNL1 | |||
|---|---|---|---|---|
| protein | ASP189.A | CYS191.A | GLY148.A | GLY219.A |
| interaction | HBDonor | Hydrophobic | HBDonor | HBDonor |
| Frame | ||||
| 0 | (None, None) | (19, 0) | (None, None) | (12, 0) |
| 1 | (11, 0) | (None, None) | (None, None) | (10, 0) |
| 2 | (None, None) | (15, 0) | (None, None) | (12, 0) |
| 3 | (25, 0) | (None, None) | (None, None) | (24, 0) |
| 4 | (25, 0) | (None, None) | (None, None) | (24, 0) |
| 5 | (None, None) | (None, None) | (None, None) | (None, None) |
| 6 | (25, 0) | (None, None) | (None, None) | (24, 0) |
| 7 | (None, None) | (15, 0) | (25, 0) | (None, None) |
| 8 | (None, None) | (None, None) | (None, None) | (None, None) |
| 9 | (None, None) | (None, None) | (None, None) | (None, None) |
[25]:
net = LigNetwork.from_ifp(results_df,lig_suppl[0],kind="frame", frame=0,rotation=270)
net.display()
[25]:
6. Docking with LeDock
LeDock is designed for fast and accurate flexible docking of small molecules into a protein. It achieves a pose-prediction accuracy of greater than 90% on the Astex diversity set and takes about 3 seconds per run for a drug-like molecule. It has led to the discovery of novel kinase inhibitors and bromodomain antagonists from high-throughput virtual screening campaigns. It directly uses SYBYL Mol2 format as input for small molecules.
6.1. Receptor preparation
In LeDock, the input file for a receptor is a PDB file with explicit hydrogens in all residues. LePro was created as a tool for preparing protein structures for docking with LeDock. Thus, at this stage, we can use the file ater sanitization steps as well as a protein structure from Jupyter Dock’s fix_protein() function.
6.2. Ligand preparation
As previously stated, the input format for ligand in LeDock is MOL2. Similarly to the receptor preparation, we can use the ligand directly after sanitization.
6.3. Docking box definition
This step can be completed in the same manner as the AutoDock Vina box definition. To obtain the identical box from AutoDock Vina docking but in LeDock format, the user only needs to change the parameter “software” from “vina” to “ledock.”
Info: The implementation of the get_box() function allows for the easy replication of binding sites between AutoDock Vina and LeDock, with the goal of replicating and comparing results between both programs.
[26]:
cmd.load(filename='1AZ8_clean_H.pdb',format='pdb',object='prot')
cmd.load(filename='1AZ8_lig_H.mol2',format='mol2',object='lig')
X,Y,Z= getbox(selection='lig',extending=5.0,software='ledock')
cmd.delete('all')
print(X,'\n',Y,'\n',Z)
{'minX': 19.57430076599121, 'maxX': 44.143798828125}
{'minY': 4.285799980163574, 'maxY': 22.409099578857422}
{'minZ': 8.378700256347656, 'maxZ': 25.75309944152832}
6.4. Docking
To run LeDock, a configuration (commonly known as dock.in) file containing all docking parameters as well as information about the receptor and ligand(s) to dock is required. Jupyter Dock uses the function generate ledock file() to generate the configuration file automatically. After configuring the parameters, the user will receive a configuration file as well as a file containing a list of ligand(s) to dock (commonly named ligand.list). After this, docking would be as simple as launching the LeDock executable and the configuration file as parameter.
generate_ledock_file( params )
params:
receptor: str or path-like string ; protein file for docking including hydrogens, format must be pdb
x: 2 element list of floats [ float , float ]; Xmin and Xmax coordinates of docking box
y: 2 element list of floats [ float , float ]; Ymin and Ymax coordinates of docking box
z: 2 element list of floats [ float , float ]; Zmin and Zmax coordinates of docking box
n_poses: float ; n_of poses to retrieve from docking
rmsd: float ; minimum RMSD diference between docking poses
l_list: _list of n strings or path-like strings [ lig1, lig2, lig3 … ] ; list of ligands or ligands paths to dock
l_list_outfile: str or path-like string ; filename to save the ligand list, needed for ledock to locate ligands
out: str or path-like string ; outfile to save docking paramemeters, needed to launch the docking
[27]:
generate_ledock_file(receptor='1AZ8_clean_H.pdb',x=[X['minX'],X['maxX']],
y=[Y['minY'],Y['maxY']],
z=[Z['minZ'],Z['maxZ']],
n_poses=10,
rmsd=1.0,
l_list='1AZ8_lig_H.mol2',
l_list_outfile='ledock_ligand.list',
out='dock.in')
[28]:
!../../bin/ledock_linux_x86 #Launch this cell to see parameters
************************************************************
* LeDock v1.0 *
* Molecular Docking Software *
* Copyright 2013-14 (C) H. Zhao PhD *
* For academic use only *
* www.lephar.com *
************************************************************
--------Usage:
--------ledock config.file !docking
--------ledock -spli dok.file !split into separate coordinates
[29]:
!../../bin/ledock_linux_x86 {'dock.in'}
6.5. DOK results file conversion to SDF
The end result of LeDock is a file with the dok extension that contains the docking properties in the same way that a pdb file does. Nonetheless, the dok file is not a widely used format for representing chemical structures. As a result, Jupyter Dock can convert dok files to the widely used sdf format. Jupyter Dock, like in the pdbqt to sdf conversion, will preserve the chemical features and save the “Pose” and “Score” results as molecule attributes.
dok_to_sdf ( params )
params:
dok_file: str or path-like ; dok file from ledock docking
output: str or path-like ; out file from ledock docking, extension must be sdf
[30]:
dok_to_sdf(dok_file='1AZ8_lig_H.dok',output='1AZ8_lig_ledock_out.sdf')
6.6. 3D visualization of docking results
As with the system visualization (section 3), the docking results can be inspected and compared to the reference structure (if exist). The ligand’s “Pose” and “Score” information will also be displayed to show how acces to this molecule’s attributes.
[31]:
view = py3Dmol.view()
view.removeAllModels()
view.setViewStyle({'style':'outline','color':'black','width':0.1})
view.addModel(open('1AZ8_clean_H.pdb','r').read(),format='pdb')
Prot=view.getModel()
Prot.setStyle({'cartoon':{'arrows':True, 'tubes':True, 'style':'oval', 'color':'white'}})
view.addSurface(py3Dmol.VDW,{'opacity':0.6,'color':'white'})
view.addModel(open('1AZ8_lig_H.mol2','r').read(),format='mol2')
ref_m = view.getModel()
ref_m.setStyle({},{'stick':{'colorscheme':'magentaCarbon','radius':0.2}})
results=Chem.SDMolSupplier('1AZ8_lig_ledock_out.sdf')
p=Chem.MolToMolBlock(results[0])
print('Reference: Magenta | LeDock Pose: Cyan')
print ('Pose: {} | Score: {}'.format(results[0].GetProp('Pose'),results[0].GetProp('Score')))
view.addModel(p,'mol')
x = view.getModel()
x.setStyle({},{'stick':{'colorscheme':'cyanCarbon','radius':0.2}})
view.zoomTo()
view.show()
Reference: Magenta | LeDock Pose: Cyan
Pose: 1 | Score: -9.06
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension:
jupyter labextension install jupyterlab_3dmol
6.7. 2D interaction table and map
Inspecting the molecular interactions is one of the most common analyses performed by a computational scientist following a docking experiment. As a result, Jupyter Dock uses ProLif to create a table of ligand-protein molecular interactions as well as the corresponding 2D map.
Info: ProLif uses RDKit and MDAnalysis to map the molecular interaction between a ligand and a protein. As a result, some protein preparations, including the use of LePro, can result in errors that impede analysis. Jupyter Dock’s fix protein() function can be used to avoid such errors and provide a suitable protein structure for the analysis.
[32]:
# load protein
prot = mda.Universe("1AZ8_clean_H_fix.pdb",guess_bonds=True)
prot = plf.Molecule.from_mda(prot)
prot.n_residues
# load ligands
path = str('1AZ8_lig_ledock_out.sdf')
lig_suppl = list(plf.sdf_supplier(path))
# generate fingerprint
fp = plf.Fingerprint()
fp.run_from_iterable(lig_suppl, prot)
results_df = fp.to_dataframe(return_atoms=True)
results_df
[32]:
| ligand | UNL1 | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| protein | ASN143.A | ASN97.A | ASP189.A | CYS191.A | CYS220.A | GLN192.A | GLY148.A | ... | SER146.A | SER147.A | SER190.A | SER214.A | THR149.A | TRP215.A | VAL213.A | ||||||
| interaction | HBAcceptor | HBDonor | Hydrophobic | HBDonor | HBDonor | Hydrophobic | Hydrophobic | Hydrophobic | Hydrophobic | HBDonor | ... | Hydrophobic | HBDonor | Hydrophobic | HBDonor | Hydrophobic | Hydrophobic | Hydrophobic | Hydrophobic | PiStacking | Hydrophobic |
| Frame | |||||||||||||||||||||
| 0 | (None, None) | (None, None) | (None, None) | (None, None) | (None, None) | (24, 9) | (0, 2) | (7, 9) | (2, 2) | (None, None) | ... | (21, 4) | (27, 5) | (21, 2) | (None, None) | (0, 4) | (13, 4) | (None, None) | (0, 2) | (None, None) | (0, 8) |
| 1 | (3, 11) | (None, None) | (21, 9) | (None, None) | (32, 11) | (24, 9) | (0, 2) | (7, 9) | (2, 2) | (28, 6) | ... | (21, 4) | (None, None) | (21, 4) | (34, 9) | (0, 4) | (None, None) | (None, None) | (0, 2) | (None, None) | (0, 8) |
| 2 | (None, None) | (None, None) | (None, None) | (None, None) | (32, 10) | (24, 9) | (0, 2) | (7, 9) | (2, 2) | (None, None) | ... | (21, 4) | (28, 5) | (21, 2) | (None, None) | (0, 4) | (None, None) | (None, None) | (0, 4) | (None, None) | (0, 8) |
| 3 | (None, None) | (27, 10) | (None, None) | (None, None) | (None, None) | (24, 9) | (0, 2) | (7, 9) | (4, 2) | (None, None) | ... | (21, 4) | (28, 0) | (21, 4) | (33, 9) | (0, 4) | (None, None) | (None, None) | (0, 2) | (None, None) | (0, 8) |
| 4 | (None, None) | (None, None) | (None, None) | (None, None) | (32, 11) | (24, 9) | (0, 2) | (7, 9) | (4, 2) | (27, 6) | ... | (21, 4) | (None, None) | (21, 4) | (None, None) | (0, 4) | (2, 4) | (None, None) | (0, 2) | (None, None) | (0, 8) |
| 5 | (3, 11) | (None, None) | (None, None) | (None, None) | (33, 11) | (24, 9) | (0, 2) | (7, 9) | (2, 2) | (27, 6) | ... | (18, 4) | (None, None) | (21, 4) | (None, None) | (0, 4) | (2, 4) | (None, None) | (0, 2) | (None, None) | (0, 8) |
| 6 | (1, 11) | (None, None) | (None, None) | (None, None) | (32, 11) | (24, 9) | (0, 2) | (7, 9) | (2, 2) | (27, 6) | ... | (21, 4) | (None, None) | (21, 4) | (None, None) | (0, 4) | (None, None) | (None, None) | (0, 2) | (None, None) | (0, 8) |
| 7 | (1, 11) | (None, None) | (None, None) | (None, None) | (None, None) | (24, 9) | (0, 2) | (7, 9) | (2, 2) | (27, 6) | ... | (21, 4) | (None, None) | (21, 4) | (32, 9) | (0, 4) | (13, 4) | (None, None) | (0, 2) | (None, None) | (0, 8) |
| 8 | (1, 11) | (None, None) | (None, None) | (None, None) | (None, None) | (24, 9) | (0, 2) | (7, 9) | (2, 2) | (None, None) | ... | (None, None) | (27, 5) | (None, None) | (None, None) | (0, 4) | (None, None) | (21, 6) | (0, 2) | (None, None) | (0, 8) |
| 9 | (None, None) | (None, None) | (None, None) | (28, 5) | (32, 11) | (24, 9) | (0, 2) | (10, 9) | (None, None) | (None, None) | ... | (None, None) | (None, None) | (None, None) | (34, 9) | (0, 4) | (2, 4) | (None, None) | (0, 2) | (12, 12) | (0, 8) |
10 rows × 30 columns
[33]:
net = LigNetwork.from_ifp(results_df,lig_suppl[0],kind="frame", frame=0,rotation=270)
net.display()
[33]: